


Bs Trần Lâm - Khoa Nội tim mạch
I. PROTEIN PHẢN ỨNG C ĐỘ NHẠY CAO (hs-CRP) LÀ GÌ?
Bên cạnh các yếu tố nguy cơ kinh điển, vai trò của các chỉ điểm viêm trong bệnh lý động mạch vành ngày càng được quan tâm. Trong số đó, protein phản ứng C đã được nhiều nghiên cứu trên thế giới đánh giá là một “chỉ điểm vàng” của quá trình viêm trong các giai đoạn khác nhau của bệnh. Hiện nay, bằng những phương pháp có độ nhạy cao, người ta có thể đo được nồng độ protein phản ứng C ở những mức rất thấp (< 0,2 mg/L), tạm dịch là protein phản ứng C độ nhạy cao (high sensitivity C-Reactive Protein, hs -CRP). Năm 2000, một khảo sát tiến hành ở Hoa kỳ đã phát hiện 12 triệu người lớn có nồng độ lipid máu không tăng nhưng nồng độ hs-CRP cao > 3 mg/L. Nhiều nghiên cứu cho thấy gần một nửa biến cố mạch vành xảy ra ở những người không tăng lipid máu, và tăng hs-CRP mạn tính làm tăng nguy cơ tử vong hoặc nhồi máu cơ tim tới 40%.
Bằng chứng về khả năng dự đoán một số kết cục tim mạch và về những liên quan bệnh lý trực tiếp gợi ý rằng việc hạ thấp hs-CRP có thể có những ảnh hưởng quan trọng trong việc đảo ngược tiến trình viêm và phòng ngừa những biến cố tim mạch tương lai [43], [47], [55], [61], [93], [100], [120].II. CƠ CHẾ BỆNH SINH CỦA XƠ VỮA ĐỘNG MẠCH VÀ BỆNH ĐỘNG MẠCH VÀNH
Nguyên nhân chủ yếu của bệnh động mạch vành (ĐMV) là do xơ vữa động mạch (XVĐM). XVĐM là hiện tượng xơ hoá thành động mạch bao gồm các động mạch trung bình và động mạch lớn. Biểu hiện chủ yếu là sự lắng đọng mỡ và các mảng tế bào ở lớp áo trong của thành động mạch gọi là mảng vữa.
Quan điểm hiện nay cho rằng XVĐM là một bệnh viêm. Đây là bệnh lý động học và tiến triển, xuất phát từ sự kết hợp của rối loạn chức năng nội mạc (RLCNNM) và quá trình viêm, hay nói một cách khác, XVĐM là một bệnh viêm mạn tính mà trong đó những cơ chế miễn dịch tương tác với những yếu tố nguy cơ để hoạt hoá những tổn thương trong cây động mạch.
II.1. Chức năng nội mạc bình thường
Nội mạc mạch máu không chỉ là một hàng rào cơ học được lát bởi một lớp đơn bào ngăn cách dòng máu và thành mạch mà còn điều hoà những chức năng quan trọng khác của giường mạch. Chức năng nội mạc bình thường quan trọng không chỉ trong việc điều hoà trương lực mạch máu mà còn trong việc tạo nên một bề mặt nội mạc không sinh huyết khối, dự phòng phản ứng viêm và hiện tượng tăng sinh thành mạch. Nội mạc bình thường được cung cấp một số cơ chế chống huyết khối, bao gồm hoạt động chống tiểu cầu của nitric oxide (NO), prostacyclin, ADPase/CD39, hoạt động chống đông của heparin, protein C, protein S, và hoạt động tiêu sợi huyết của yếu tố hoạt hoá plasminogen mô (tissue Plasminogen Activator, tPA).
Nội mạc mạch máu có thể nhạy cảm với những thay đổi huyết động và những tín hiệu phát sinh từ dòng máu. Để kiểm soát trương lực vận mạch, nội mạc tổng hợp và phóng thích những chất hoạt mạch như prostacyclin, yếu tố tăng phân cực (hypepolarizing factor), endothelin..., và quan trọng nhất là NO. Sự hằng định nội mô của mạch máu được duy trì bởi một sự cân bằng giữa các yếu tố co mạch và giãn mạch có nguồn gốc nội mạc này. Khi sự cân bằng này bị vỡ qua trung gian của những yếu tố viêm và những yếu tố nguy cơ kinh điển, giường mạch trở nên nhạy cảm với sự hình thành mảng vữa.
Các yếu tố giãn mạch được phân thành 2 nhóm:
* Các chất giãn mạch độc lập với nội mạc tác động trực tiếp lên cơ trơn thành mạch bất kể tình trạng của nội mạc như thế nào, và
* Các chất giãn mạch phụ thuộc nội mạc bắt buộc cần có sự hiện diện của một nội mạc còn hoạt động bình thường.
* Nitric oxide (NO): Năm 1980, Furchgott và Zawadzki nhận thấy sự hiện diện của tế bào nội mạc (TBNM) mạch máu là cần thiết để acetylcholine có tác dụng giãn mạch. Nếu nội mạc bị loại bỏ, mạch máu sẽ không giãn với acetylcholine, nhưng vẫn đáp ứng với glyceryl trinitrate. Sự giãn mạch phụ thuộc nội mạc này của cơ trơn mạch máu với acetylcholine thông qua vai trò của một yếu tố trung gian nội sinh. Lúc đầu, yếu tố này được đặt tên là yếu tố giãn mạch có nguồn gốc nội mạc (Endothelium Derived Relaxing Factor, EDRF), về sau, nó được xác định là NO. NO là yếu tố giãn mạch chủ yếu, đóng một vai trò quan trọng trong sự điều hoà trương lực mạch máu và chức năng vận mạch. Ngoài hiệu quả giãn mạch, NO còn có tác dụng chống viêm, chống sinh xơ vữa, đó là:
* NO ức chế sự kết dính của bạch cầu vào nội mạc.
* NO ức chế sự tăng sinh và di chuyển của tế bào cơ trơn (TBCT).
* NO ức chế sự hoạt hoá và bộc lộ của những phân tử kết dính.
* NO ức chế sự ngưng tập của tiểu cầu.
* NO là một chất chống oxy hoá .
Về mặt sinh lý, gắng sức là một kích thích cơ học quan trọng đối với nội mạc qua trung gian áp lực xé (shear stress) do tăng dòng máu. Khi nội mạc bị kích thích bởi áp lực xé do tăng dòng chảy, NO synthase của nội mạc (endothelial NO synthase - eNOS) được hoạt hoá thông qua một con đường không phụ thuộc thụ thể bằng cách phosphoryl hoá NO synthase, và sản xuất NO từ tiền chất của nó là L- arginine. Ngược lại, bradykinin hoặc serotonin có nguồn gốc từ tiểu cầu kích thích nội mạc qua những thụ thể đặc hiệu dẫn đến hoạt hoá NO synthase qua trung gian protein G. Sau khi được tổng hợp, NO khuyếch tán qua màng của TBNM và đi vào TBCT mạch máu, ở đó, nó hoạt hoá guanylate cyclase dẫn đến tăng nồng độ nội bào của guanosine monophosphat vòng (cGMP). cGMP đóng vai trò trung gian cho nhiều tác dụng sinh học của NO, bao gồm sự kiểm soát trương lực mạch máu và chức năng tiểu cầu. Nội mạc không chỉ có vai trò trong giãn mạch mà còn là nơi sản xuất các yếu tố co mạch, chủ yếu là endothelin-1.
Trước khi xuất hiện mảng xơ vữa, chức năng giãn mạch của nội mạc đã sớm bị suy giảm.
II.2. Rối loạn chức năng nội mạc trong xơ vữa động mạch và bệnh mạch vành
Rối loạn chức năng nội mạc (RLCNNM) là một thuật ngữ rộng bao hàm giảm sản xuất hoặc giảm độ khả dụng sinh học của NO và / hoặc một sự mất cân bằng giữa các yếu tố giãn mạch và co mạch có nguồn gốc nội mạc.
II.2.1. Giảm hoạt động sinh học của NO
Sự giãn mạch phụ thuộc nội mạc của ĐMV bị suy giảm trong bệnh lý XVĐM dẫn đến sự co mạch nghịch thường, có thể dẫn đến giảm tưới máu cơ tim và thiếu máu cục bộ (TMCB) cơ tim. Tuy nhiên, RLCNNM có thể xảy ra trước khi có những biểu hiện cấu trúc của XVĐM, chẳng hạn, sự đáp ứng bất thường đối với acetylcholine được phát hiện ngay cả ở những ĐMV mà trên chụp mạch còn bình thường. Như vậy, RLCNNM có thể là một yếu tố dự đoán (YTDĐ) độc lập những biến cố tim mạch tương lai.
Trong khi đáp ứng với những yếu tố nguy cơ (YTNC) tim mạch kinh điển như tăng cholesterol máu, hút thuốc lá, THA, đái tháo đường (ĐTĐ)..., khả năng bảo vệ nội sinh của nội mạc mạch máu bắt đầu bị suy giảm. Trong những giai đoạn sớm của XVĐM, tăng cholesterol máu kích thích bạch cầu kết dính vào nội mạc, LDL cholesterol (LDL-C) tập trung trong thành mạch và được oxy hoá. Sự oxy hoá là cần thiết cho cuộc sống, tuy nhiên, nếu quá trình này vượt ra khỏi tầm kiểm soát nó sẽ trở nên có hại cho giường mạch. LDL đã oxy hoá (oxLDL) gây hoạt hoá và làm thay đổi những đặc điểm sinh học của nội mạc phần nào bằng cách làm giảm nồng độ NO nội bào. Những gốc tự do dẫn xuất từ oxy có khả năng bất hoạt nhanh chóng NO thành peroxitrite (ONOO-) không còn những chức năng giãn mạch và bảo vệ mạch máu của NO nữa. Nguồn gốc của những anion superoxide là xanthine oxidase, và đặc biệt là NADH/NADPH oxidase sản xuất những anion superoxide trong đại thực bào (ĐTB) sau khi có sự kích thích của angiotensin II, đây là một chất trung gian chủ yếu của stress oxy hoá trong thành mạch. Trong những giai đoạn sớm của bệnh ĐMV, những gốc oxy phản ứng là do nội mạc phóng thích; tuy nhiên, trong quá trình XVĐM sau này khi thành XVĐM dày lên, ĐTB sẽ phóng thích những anion superoxide tích luỹ trong lớp áo trong, và tiến trình tiền oxy hoá lan rộng đến thành mạch.
Ngoài sự bị huỷ hoại do những gốc oxy phản ứng, hoạt động sinh học của NO có thể bị giảm thông qua những cơ chế khác như giảm sản xuất NO. Chẳng hạn, trong tăng cholesterol máu có một sự tác động qua lại giữa oxLDL và vài quá trình dẫn truyền tín hiệu qua trung gian thụ thể dẫn đến giảm tổng hợp NO bởi eNOS. Thực nghiệm cho thấy trong XVĐM hoạt động của eNOS bị giảm. Tuy nhiên, tổng sản xuất NO có thể tăng bởi vì NO không chỉ sản xuất bởi eNOS mà còn bởi NO synthase của tế bào thần kinh (neuronal NO synthase, nNOS), và quan trọng hơn là bởi iNOS (inducible NO synthase) của ĐTB và những loại tế bào khác trong mảng xơ vữa.
II.2.2. Những cơ chế cơ bản của sự hoạt hoá nội mạc
Sự biến đổi nội mạc không chỉ gây nên rối loạn chức năng giãn mạch của mạch máu mà còn dẫn đến một số quá trình hoạt hoá nội mạc có những ảnh hưởng quan trọng, đó là quá trình viêm, sự tăng sinh và hiện tượng chết theo chương trình của tế bào mạch máu.
* Quá trình viêm: Giảm hoạt động sinh học của NO hoặc tăng stress oxy hoá dẫn đến sự nitrô hoá tyrosine của những protein trong thành mạch và hoạt hoá yếu tố nhân B (Nuclear Factor-B, NF-B). NF-B là một protein sao chép làm gia tăng sự tăng sinh của TBCT và những tế bào khác. Hơn nữa, giảm hoạt động sinh học của NO cùng với tăng stress oxy hoá sẽ kích thích sự sản xuất những cytokine như là những interleukin (IL), yếu tố hoại tử u-a (tumor necrosis factor-a, TNF-a), protein hoá hướng động tế bào đơn nhân-1 (monocyte chemoattractant protein-1, MCP-1) hoặc interferon (IFN), do vậy hấp dẫn tế bào đơn nhân (TBĐN). Angiotensin II có thể kích thích sản xuất những gốc oxy phản ứng, gia tăng sự bộc lộ của những cytokin tiền viêm như IL-6, MCP-1, và điều hoà lên phân tử kết dính tế bào mạch máu-1 (vascular cell adhesion molecule-1,VCAM-1,) ở TBNM. Những YTNC mới hơn như tăng nồng độ hs-CRP cũng có thể thúc đẩy RLCNNM bằng cách kiềm hãm sự sản xuất và làm giảm hoạt tính sinh học của NO. Những biến đổi này của nội mạc thúc đẩy hiện tượng viêm trong thành mạch, khởi đầu và tiến triển của một tổn thương xơ vữa.
* Sự tăng sinh thành mạch: Sự mất cân bằng oxy hoá - khử giữa NO và các gốc oxy phản ứng trực tiếp tương tác với sự bộc lộ của các gen có liên quan đến quá trình tăng sinh thành mạch. Trong khi NO làm giảm sự tăng sinh của TBCT thành mạch và sự di chuyển của TBĐN thì ET-1 và angiotensin II lại có những tác dụng tiền xơ vữa trực tiếp trên thành mạch. Nghiên cứu thực nghiệm cho thấy những con chuột thiếu eNOS bị giảm sự giãn mạch phụ thuộc nội mạc và tăng quá mức sự tăng sinh lớp áo trong sau một kích thích bên ngoài thành mạch .
Một quá trình quan trọng đáp ứng đối với sự xuất hiện mảng xơ vữa là quá trình tái cấu trúc mạch máu (vascular remodelling). Đây là sự gia tăng kích thước mạch máu trong giai đoạn sớm của XVĐM để bù trừ cho sự xâm lấn lòng mạch của mảng xơ đang phát triển. Quá trình này là hậu quả của tăng dòng máu ở vị trí mảng xơ đang phát triển. Dòng máu bị giảm mạn tính liên quan với giảm đường kính mạch máu; trong khi đó, một sự gia tăng dòng máu, và tất nhiên cả tăng áp lực xé trên thành mạch sẽ dẫn đến sự gia tăng kích thước mạch máu. Gần đây, dựa vào những nghiên cứu ở chuột, người ta cho rằng NO có nguồn gốc từ iNOS chịu trách nhiệm cho sự tăng trưởng thành mạch khởi đầu quá trình tái cấu trúc, trong khi đó, eNOS liên quan với sự ức chế hiện tượng dày lên của thành mạch. Tuy nhiên, cơ chế tái cấu trúc mạch máu thích nghi bù trừ ban đầu này có thể trở nên có hại trong những giai đoạn tiến triển của bệnh với sự gia tăng tính kém ổn định của mảng xơ.
* Hiện tượng chết tế bào theo chương trình: Sự mất cân bằng oxy hoá - khử giữa NO và các gốc oxy phản ứng cũng kiểm soát hiện tượng chết tế bào theo chương trình, có thể làm biến đổi những chức năng sinh lý khác nhau của lớp TBNM. Các YTNC tim mạch ảnh hưởng đến sự cân bằng của các tế bào mạch máu. Chẳng hạn, oxLDL hoặc angiotensin II thúc đẩy hiện tượng chết theo chương trình của TBNM; ngược lại, áp lực xé, NO nội mạc và các vitamin chống oxy hoá lại ức chế hiện tượng này.
II.3. Vai trò của viêm trong xơ vữa động mạch và bệnh động mạch vành
XVĐM là một bệnh viêm mạn tính. Trong quá trình này, các chất trung gian viêm đóng một vai trò chủ yếu trong sự khởi đầu, tiến triển, và cuối cùng là vở mảng xơ vữa.
Những bằng chứng hiện nay cho thấy hs-CRP - một chỉ điểm viêm hệ thống - là một trong những yếu tố dự đoán (YTDĐ) độc lập, mạnh nhất của biến cố tim mạch ở người khoẻ mạnh cũng như ở bệnh nhân (BN) có cơn đau thắt ngực ổn định hoặc không ổn định, BN sau nhồi máu cơ tim. Sự gia tăng nồng độ của hs-CRP là một YTDĐ độc lập nguy cơ NMCT, đột quỵ, bệnh động mạch ngoại biên, đột tử do tim, và sự tái hẹp sau can thiệp mạch vành qua da (CTMVQD).
II.3.1. CRP- một protein chính để đánh giá quá trnh viêm
* Nguồn gốc và cấu tạo: Năm 1930, tại Viện Nghiên cứu Y học Rockefeller, Tillett và Francis đã phát hiện trong huyết thanh của BN bị viêm phổi do phế cầu có một loại protein có khả năng kết tủa với C-polysaccharide của vỏ phế cầu khuẩn và đặt tên nó là protein phản ứng C (C-Reactive Protein, CRP). Có khoảng trên 70 bệnh lý bao gồm nhiễm trùng cấp, nhiễm trùng mạn, nhiễm vius, và các bệnh lý không nhiễm trùng như THA, béo phì, hút thuốc lá, ĐTĐ, hội chứng đề kháng insulin, bệnh ĐMV, bệnh tự miễn, bệnh ác tính ...gây tăng nồng độ CRP.
CRP được tổng hợp chủ yếu tại gan dưới tác dụng kích thích của các cytokin viêm như IL-6, IL-1, và IFN-a) khi cơ thể đang có hiện tượng viêm. Ngoài gan ra, mô mỡ cũng đóng một vai trò quan trọng trong sản xuất CRP. CRP cũng được sản xuất bởi TBCT trong ĐMV, nó hiện diện ở những mảng xơ vữa. Renu Virmani và cs (2002) nghiên cứu giải phẫu bệnh ĐMV của 302 BN bị chết, trong đó, 73 BN chết do vỡ hoặc xói lở mảng xơ vữa, 73 BN có mảng xơ vữa ổn định, và nhóm chứng bao gồm 158 trường hợp đột tử không do nguyên nhân tim mạch. Kết quả cho thấy mức hs-CRP trung bình ở mảng xơ vữa bị vỡ là 3,2 mg/L, 2,9 mg/L ở mảng xơ vữa bị xói lở, 2,5 mg/L ở mảng xơ vữa ổn định, và chỉ 1,4 mg/L ở nhóm chứng.
CRP có trọng lượng phân tử là 120.000 kDA, được cấu tạo bởi 5 chuỗi polypeptid kết hợp không chặt chẽ, sắp xếp đối xứng chung quanh lỗ trung tâm. Theo Khreiss và cs (2004), CRP phải trải qua một sự thay đổi cấu trúc ở vị trí gắn vào màng TBNM, từ dạng 5 chuỗi polypeptid chuyển thành dạng 1 chuỗi. Dạng CRP 1 chuỗi có thể gây hoạt hoá nội mạc trong vòng 4 giờ, trong khi đó, để có được tác dụng tương tự dạng CRP 5 chuỗi phải cần đến 24 giờ, bởi vì sự hoạt động của CRP 1 chuỗi dựa vào môi trường mô hơn là môi trường huyết thanh (hình 1.1).
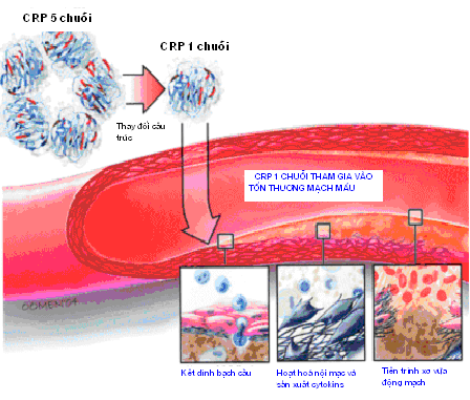
Hình 1.1. Mô hình cấu tạo và hoạt động của CRP.
(Nguồn: Subodh Verma et al (2004), “CRP: Structure affects function”, Circulation, 109, pp. 1914-17).
Gen điều hành sự tổng hợp CRP nằm trên nhánh ngắn của nhiễm sắc thể số 1. Tính di truyền của nồng độ hs-CRP cơ bản thể hiện ở xấp xỉ 40% những nghiên cứu gia đình. Hiện nay, người ta đã tìm thấy 3 dạng gen của CRP liên quan với những thay đổi của nồng độ hs-CRP. Có lẽ, những BN nguy cơ cao có một dạng gen gây giảm tính ổn định của cấu trúc CRP 5 chuỗi, do vậy thúc đẩy sự hình thành cấu trúc CRP 1 chuỗi, hay nói một cách khác, họ có một dạng gen làm gia tăng tính ổn định của cấu trúc 1 chuỗi gắn vào màng tế bào, từ đó dẫn đến tăng hoạt hoá TBNM.
Nồng độ hs-CRP không bị ảnh hưởng bởi tuổi, giới, và nhịp ngày đêm. Hơn nữa, CRP có tính ổn định cao cho phép đo lường dễ dàng, chính xác, và cho những kết quả giống nhau từ những mẫu huyết tương tươi, lưu trữ, hoặc đông lạnh. Điều này phần nào do CRP có thành phần pentraxin ổn định, và có thời gian nửa đời dài 18-20 giờ. Ở người bình thường, nồng độ của CRP hoàn toàn ổn định và thường < 1 mg/L.
* CRP và đáp ứng pha cấp: Pha cấp liên quan đến những biến đổi sinh lý và chuyển hoá diễn ra ngay sau nhiễm trùng hay tổn thương mô, và có ảnh hưởng đến toàn thân gọi là đáp ứng pha cấp. Khác với tính đặc hiệu của miễn dịch thể dịch và miễn dịch tế bào, các thay đổi của pha cấp không đặc hiệu và xảy ra trong nhiều loại bệnh lý. Đáp ứng này tạo ra sự thay đổi về nồng độ của các protein huyết tương. Một protein được gọi là protein pha cấp chỉ khi nồng độ của nó trong huyết tương tăng hoặc giảm đi ít nhất là 25% trong vòng 7 ngày kể từ khi có tổn thương mô hay viêm nhiễm.
CRP được xem là một trong hơn 50 protein pha cấp chính vì đạt được tiêu chuẩn trên, đồng thời có tính nhạy cảm cao và nồng độ huyết tương của nó thay đổi rất sát với quá trình viêm; vì vậy, nó phản ánh và đánh giá chinh xác quá trình viêm. Sau khi khởi phát viêm hay tổn thương mô cấp, sự tổng hợp CRP tăng lên trong vòng 6 - 12 giờ và tăng gấp đôi mỗi 8 giờ, đạt đến đỉnh cao sau 36 - 50 giờ và trở về bình thường sau 5 - 7 ngày nếu không còn tác nhân kích thích.
Ở các nhiễm trùng do vi khuẩn, nồng độ CRP thường >100 mg/L. Với những trường hợp sốt không rõ nguyên nhân, CRP tăng cao là một chỉ điểm tốt cho nhiễm vi khuẩn. Mức tăng CRP trong nhiễm virus thường không vượt quá 40 mg/L. Trong các bệnh lý viêm không do nhiễm trùng CRP thường <50 mg/L nếu không có tổn thương mô trầm trọng. CRP cũng tăng trong các bệnh lý khác như béo phì, THA, ĐTĐ, hội chứng đề kháng insulin, XVĐM, bệnh ĐMV...Nó phản ánh tình trạng viêm mạn tính trong các bệnh lý này.
II.3.2. Cơ chế hoạt động của CRP
Viêm động mạch là nguồn gốc dẫn đến quá trình huyết khối xơ vữa. Kết quả của nhiều nghiên cứu cho thấy CRP có một vai trò bệnh sinh trực tiếp trong sự hoạt hoá nội mạc mạch máu, qúa trình viêm và tổn thương XVĐM (hình 1.2).
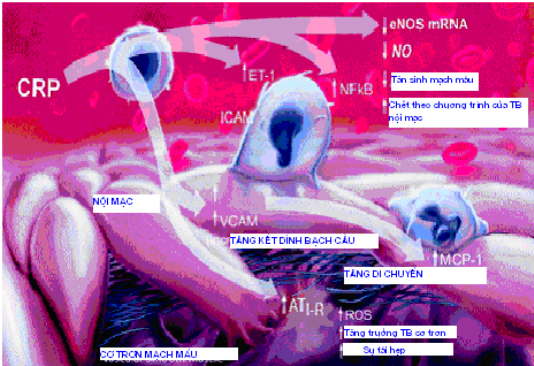
Hình 1.2. Vai trò của CRP trong rối loạn chức năng nội mạc, viêm và xơ vữa động mạch.
(Nguồn: Paul E. Szmitko et al (2003), “New markers of inflammation and endothelial cell activation”, Circulation,108, pp.1917-27).
* CRP có thể điều hoà xuống sự sao chép của eNOS ở TBNM và làm rối loạn eNOSmRNA với hậu quả là làm giảm cả NO cơ bản và kích thích. CRP liên quan với RLCNNM ở BN bị bệnh ĐMV là do làm tăng stress oxy hoá và làm giảm độ khả dung sinh học của NO. CRP cũng làm giảm phóng thích prostacyclin từ TBNM, kích thích TBĐN phóng thích ET-1.
Nghiên cứu cho thấy nồng độ hs-CRP tương quan nghịch với sự tổng hợp NO cơ bản của nội mạc. Hơn nữa, ở BN bị bệnh ĐMV, nồng độ CRP tăng liên quan với sự suy giảm tính phản ứng của nội mạc động mạch cẳng tay, và sự bình thường hoá nồng độ hs-CRP liên quan với một sự cải thiện có ý nghĩa chức năng nội mạc.
CRP không chỉ là một chỉ điểm của sự hoạt hoá nội mạc mà còn chủ động đóng vai trò bệnh sinh của viêm ngay tại giường mạch:
* CRP có thể hoạt động trực tiếp như một kích thích tiền viêm đối với ĐTB bằng cách gắn vào những thụ thể đặc hiệu FcgRII trên ĐTB dẫn đến hiện tượng hoá ứng động của TBĐN, giúp TBĐN xâm nhập vào trong thành mạch.
* CRP kích thích TBĐN phóng thích IL-1b, IL-6 và TNF-a.
* CRP điều hoà lên những phân tử kết dính (ICAM-1 và VCAM-1), MCP-1 và E-selectin trên TBNM mạch vành, và opsonin hoá LDL-C tạo thuận lợi cho sự thu nhận LDL-C của ĐTB
* CRP có khả năng làm cho TBNM dễ bị huỷ hoại bởi tế bào T CD4+ độc tế bà.
* Những dữ liệu gần đây cho thấy mô động mạch có thể sản xuất CRP và những protein bổ thể. Những sản phẩm này và mRNA liên quan của chúng được điều hoà lên đáng kể trong mảng xơ vữa. CRP hoạt hoá hệ thống bổ thể, kết hợp với phức hợp tấn công màng trong những tổn thương xơ vữa sớm làm gia tăng hơn nữa quá trình viêm trong thành mạch.
* Gần đây hơn, S. Verma, R. P. Palusinski và cs phát hiện CRP có thể tạo thuận lợi cho TBNM chết theo chương trình, gia tăng sự hoạt hoá TBNM gây nên bởi CD14.
* Những kết quả sơ bộ từ nghiên cứu của Paul E. Szmitko và cs cho thấy CRP cũng có tiềm năng điều hoà lên NF-B, đây là yếu tố nhân chủ yếu tạo thuận lợi cho sự sao chép của vài gen tiền xơ vữa. Những tác dụng tiền xơ vữa của CRP dường như cũng bị thay đổi bởi những YTNC khác và những chiến lược điều trị. Ví dụ, tăng đường máu làm gia tăng những tác động của CRP trên sự hoạt hoá của TBNM; trong khi đó, những can thiệp dược lý bằng statin, glitazone và bosentan (một tác nhân đối kháng thụ thể endothelin) làm giảm những quá trình này.
* CRP dường như cũng ức chế sự sống sót và biệt hoá của tế bào tiền sinh nội mạc có nguồn gốc từ tuỷ xương (bone marrow-derived Endothelial Progenitor Cell, EPC). Người ta cho rằng những tế bào EPC đóng một vai trò quan trọng trong quá trình tân sinh mạch máu sau sinh, và khả năng ức chế tế bào EPC của CRP có thể là một cơ chế quan trọng nhằm ức chế hiện tượng sinh mạch bù trừ trong TMCB mạn tính.
Như vậy, CRP không chỉ là một chỉ điểm viêm của XVĐM / biến cố mạch vành mà còn là một chất trung gian của bệnh bởi vì nó tham gia vào cơ chất hình thành tổn thương cơ bản, vỡ mảng xơ vữa, và tắc nghẽn mạch vành thông qua sự tương tác và làm biến đổi kiểu hình của TBNM.
* Những tác dụng tiền sinh xơ vữa của CRP không những ảnh hưởng đến nội mạc mà còn lan đến TBCT mạch máu. Bằng chứng gần đây gợi ý rằng CRP, ở nồng độ dự đoán biến cố tim mạch, trực tiếp điều hoà lên thụ thể AT1 của angiotensin ở TBCT mạch máu in vitro và in vivo, kích thích sản xuất yếu tố mô (Tissue Factor, TF), kích thích sự di chuyển và tăng sinh của TBCT mạch máu, kích thích sự hình thành lớp áo trong mới và sự sản xuất những gốc oxy phản ứng.
Tuy nhiên, Subodh Verma và cs (2004) nhận thấy CRP cũng điều hoà lên những protein ức chế bổ thể và bảo vệ TBNM khỏi những tổn thương qua trung gian bổ thể. Như vậy, một sự cân bằng giữa tác dụng tiền sinh xơ vữa và chống sinh xơ vữa của CRP trên thành mạch có thể đóng một vai trò quan trọng trong sự phát triển của XVĐM.
Tóm lại, XVĐM không còn được xem là một rối loạn lipid đơn thuần nữa. Càng ngày càng có nhiều bằng chứng chứng minh viêm là nguồn gốc của XVĐM và những biến chứng của nó. Một số cơ chế và chất trung gian của viêm đã được xác định, trong số đó, CRP là một chỉ điểm quan trọng nhất. Rõ ràng, sự hiểu biết những cơ chế và những chất trung gian của RLCNNM và viêm có thể cung cấp những cái đích mới để dự đoán, dự phòng và điều trị bệnh tim mạch.
TÀI LIỆU THAM KHẢO
- ACC/AHA (2000), “Inflammation, heart disease and stroke: The role of CRP”, Circulation, pp. 1-3.
- Allan R. Brasier et al (2002), “Vascular inflammation and the renin-angiotensin system”, Arteriosclerosis, Thrombosis, and Vascular Biology, 22(8), pp. 1257-70.
- Antonio MG et al (2000), “Blood lipids and coronary heart disease”, The ILIB lipid handbook for clinical practice, 2nd edition, pp. 7-18.
- Baohua JI (2004), “CRP and cardiovascular disease”, Journal of Geriatric Cardiology, 1(1), pp. 17-20.
- Barclay L (2003), “CRP testing and CVD risk”, Circulation, 107, pp. 499-511.
- Blake GJ, Ridker PM (2001), “Novel clinical markers of vascular wall inflammation”, Circulation, 89, pp. 763-80.
- Deepak L. Bhatt, Eric J. Tipol (2002), “Need to test the arterial inflammation hypothesis”, Circulation, 106, pp. 136.
- Derek P.Chew, Deepak L. Bhatt et al (2001), “Incremental prognostic value of elevated baseline CRP among established markers of risk in percutaneous coronary intervention”, Circulation, 104, pp. 992-98.
- Emmanouil Zouridakis et al (2004), “Markers of inflammation and rapid coronary artery disease progression in patients with stable angina pectoris”, Circulation, 110, pp.1757-53.
- Frits Haverkate et al (1997), “Production of CRP and risk of coronary events in stable and unstable angina”, The Lancet, pp. 462-466.
- Garcia X. Moll et al (2000), “CRP in patients with chronic stable angina: differences in baseline serum concentration between women and men”, Circulation, 90, pp. 742-47.
- Gavin J. Blake, Paul M. Sidker (2001), “Novel clinical markers of vascular wall inflammation”, Circulation, 89, pp. 763-74.
- Gersh BJ, Brauwald E (2001), “Chronic coronary artery disease”, Heart disease, 6th ed, CD-ROM.
- Goran K. Hansson (2005), “Inflammation, atherosclerosis, and coronary artery disease”, NEJM, 352, pp. 1685-95.
- Haidari M et al (2001), “Evaluation of CRP, a sensitive marker of inflammation, as a risk factor for stable coronary artery disease”, Clinical Biochemistry, 34(4), pp. 309-15.
- Jeffrey L. Anderson (2005), “Infection, antibiotics, and atherothrombosis - End of the road or new beginning?”, NEJM, 352 (16), pp. 1706-08.
- John Danesh et al (2000), “Low grade inflammation and coronary heart disease: prospective study and updated meta-analyses”, BMJ, 321, pp. 199-204.
- Judith Mackay, George A. Mensah (2004), “Risk factors”, “Deaths from coronary heart disease”, The atlas of heart disease and stroke, WHO, pp. 22-42, 48-49.
- Matthew J. Sorrentino et al (2004), “Inflammation and vascular disease: the role of CRP”, Journal of Geriatric Cardiology, 1(1), pp. 11-13.
- Mike Mitka (2004), “Biomarkers for coronary heart disease: Predictive value or background noise”, JAMA, 292 (23), pp. 2824-26.
- Patrick Vallance, Norman Chan (2001), “Endothelial function and nitric oxide: clinical relevance”, Heart, 85, pp. 342-350.
- Paul E. Szmitko, Chao-Hung Wang et al (2003), “New markers of inflammation and endothelial cell activation”, “Biomarkers of vascular disease linking inflammation to endothelial activation”, Circulation,108, pp. 1917-27, 2041-51.
- Peter Libby et al (2002), “Inflammation and atherosclerosis”, Circulation, 105, pp. 1135-45.
- Renu Virmani et al (2002), “Elevated CRP values and atherosclerosis in sudden coronary death”, Circulation, 105, pp. 2019-29.
- Ridker PM, Charles H. Hennekens et al (2000), “CRP and other markers of inflammation in the prediction of cardiovascular disease in women”, NEJM, 342 (12), pp. 836-843.
- Ridker PM. (2003), “Clinical application of CRP for cardiovascular disease detection and prevention”, Circulation, 107, pp. 363-70.
- Ridker PM. et al (2004), “Clinical usefullness of very high and very low levels of CRP across the full range of framingham risk scores”, Circulation, 109, pp.1955-59.
- Russel Ross (1999), “Atherosclerosis - An inflammation disease”, NEJM, 340 (2), pp. 115-126.
- 29. Samir R Kapadia, David J Moliterno (2000), “Chronic ischemic syndromes”, Manual of cardiovascular medicine, Loppincott William & Wilkins, pp. 71-95.
- Sidney C. Smith, Richard V. Milani et al (2004), “Atherosclerotic vascular disease conference”, Circulation, 109, pp. 2613-16.
- Sidney C. Smith et al (2004), “CDC/AHA workshop on markers in inflammation and cardiovascular disease”, Circulation, 110, pp. e550-e553.
- Subodh Verma et al (2004), “CRP: Structure affects function”, Circulation, 109, pp. 1914-17
- Tomai F (2004), “CRP and microvascular function”, Heart, 90, pp. 727-728.
- Umed A. Ajani et al (2004), “Prevalence of high CRP in persons with serum lipid concentrations within recommended values”, Clinical Chemistry, 50, pp.1618-22.
- Volker Schachinger et al (2002), “Atherogenesis - recent insight into basic mechanisms and their clinical impact”, Nephrol Dial Transplant, 17, pp. 2055-64.
- Wim K. Lagrand et al (1999), “CRP as a cardiovascular risk factor more than an epiphenomenon”, Circulation, 100, pp. 96-102.
- Winter RJ et al (2004), “The prognostic value of pre-procedural plasma CRP in patients undergoing elective coronary angioplasty”, Circulation, 102, pp. 126-132.
- Zebrack JS et al (2002), “CRP and angiographic coronary artery disease: independent and additive predictors of risk in subjects with angina”, JACC, 39 (4), pp. 632-37.
- 02/08/2012 10:24 - Xuất huyết vỡ tĩnh mạch trướng thực quản: Điều trị…
- 30/07/2012 21:04 - Chẩn đoán hình ảnh Ung thư tiền liệt tuyến
- 24/07/2012 14:52 - Hội chứng Brugada - một nguyên nhân của đột tử do…
- 16/07/2012 09:50 - Hướng xử trí các bất thường tế bào âm đạo cổ tử cu…
- 10/07/2012 20:22 - Hen phế quản và hút thuốc lá
- 29/06/2012 15:05 - Hội chứng Brugada - một nguyên nhân của đột tử do…
- 20/06/2012 07:13 - Truyền máu khối lượng lớn-nguy cơ và lựa chọn máu …
- 19/06/2012 19:55 - Bóc tách động mạch chủ và CT scan chẩn đoán (p.2)
- 19/06/2012 15:14 - Bóc tách động mạch chủ và CT scan chẩn đoán (p.1)
- 19/06/2012 13:04 - Hội chứng Brugada – một nguyên nhân của đột tử do …




